| CPC C12Q 1/6858 (2013.01) [C12Q 1/686 (2013.01); G16B 20/10 (2019.02); G16B 20/50 (2019.02); G16B 30/10 (2019.02); G16B 40/00 (2019.02); G06N 20/00 (2019.01); G16B 40/20 (2019.02)] | 5 Claims |
|
1. A method for determining genome instability based on massively parallel next generation sequencing (NGS), wherein, the method is not used for a disease diagnosis, but for a determination based on a bioinformatics analysis, wherein the method is used to determine whether there is a lack of homologous recombination by calculating a comprehensive value of at least one selected from the group consisting of a pathogenic point mutation or an insertion/deletion (Indel) mutation of homologous recombination repair (HRR) genes, a biallelic pathogenic mutation burden of the HRR genes, a mutational signature of the HRR genes, a copy number variation (CNV) of the HRR genes, a copy number (CN) burden of the HRR genes, and a genomic structural variation,
wherein the method specifically comprises the following steps:
1) Performing targeted sequencing on a tumor sample and a baseline sample based on the NGS, wherein the targeted sequencing comprises:
obtaining a DNA library from the tumor sample and a DNA library from the baseline sample;
designing probes for a single-nucleotide polymorphism (SNP)-panel as follows:
a) removing gaps and regions with a low mappability on the human reference genome, and removing genomic regions having a length of 200 bp and a GC content, wherein the GC content is higher than 60% and less than 30%;
b) removing regions having a length of 120 bp and comprising three or more loci that greater than 50% of an Asian population are heterozygous at; and
c) screening SNP loci in a human genomic database in regions for designing the probes, excluding the regions previously removed, for the SNP-panel as follows: determining regions with the three or more loci that greater than 50% of an Asian population are heterozygous at, determining whether Hardy-Weinberg equilibrium is reached for the SNP loci being screened other than the three or more loci that greater than 50% of the Asian population are heterozygous at, removing or retaining each SNP locus within the SNP loci based on whether they have reached Hardy-Weinburg equilibrium and whether a sequence comprising the SNP and 100 bp extended on both sides of the SNP aligns with the human reference genome at 10 or less alignment positions;
performing a hybrid capture comprising: mixing a DNA library obtained from the tumor sample to obtain a tumor DNA library mixture and mixing a DNA library obtained from the baseline sample to obtain a baseline DNA library mixture, adding human Cot-1 DNA and blocking sequences to each DNA library mixture, and then subjecting each DNA library mixture to an evaporation drying with a vacuum centrifugal concentrator to obtain evaporated resulting DNA libraries;
performing a DNA hybridization with the probes to each evaporated resulting DNA library to obtain captured libraries and amplifying the captured libraries to obtain amplified captured libraries; and
performing a library sequencing comprising: mixing the captured DNA library obtained from the tumor sample and the captured DNA library obtained from the baseline sample to obtain a resulting captured DNA library mixture, wherein the resulting captured DNA library mixture comprises a 6:1 ratio of tumor captured library to baseline captured library; and using a gene sequencer to conduct a library sequencing on the captured DNA library mixture on a computer to obtain sequencing data; after the library sequencing on the computer is completed, processing the sequencing data to obtain processed NGS sequences of the tumor sample and the baseline sample, wherein the processed NGS sequences are without adapters and primers and include HRR genes;
2) Obtaining pre-processed whole genome sequencing (WGS) data of the tumor sample and the baseline sample, and aligning the pre-processed WGS data of the tumor sample and the baseline sample and the processed NGS sequences of the tumor sample and the baseline sample with a human reference genome to obtain position information of each sequence, alignment quality information, and aligned results, analyzing the aligned results for quality evaluation, and using the aligned results to obtain a B-allele frequency (BAF) and a CN of each of a plurality of heterozygous loci in a targeted region of the tumor sample;
3) Screening out a heterozygous locus of the plurality of heterozygous loci of the tumor sample and acquiring an allele frequency of the heterozygous locus;
4) Determining allelic imbalance (AI) scores of telomeric regions and regions other than centromeres and telomeres in the genome, wherein:
a) the AI scores of the regions other than the centromeres and the telomeres are calculated as follows: using the processed NGS sequences of the tumor sample and the baseline sample determined by the targeted sequencing in step 1 to, for each of the tumor sample and baseline sample: count a coverage of each probe of the probes; using locally weighted regression (LWR) to correct the coverage of each probe; and using the corrected coverage to calculate a CN corresponding to each probe and an allele frequency of a SNP locus on each probe,
wherein calculation formulas for each probe for the baseline sample are as follows:
calculation formula for CN (cnT):
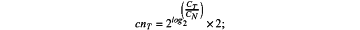 calculation formula for the allele frequency (BAF):
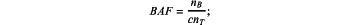 and
calculation formulas for each probe for the tumor sample with a tumor content of ρ are as follows:
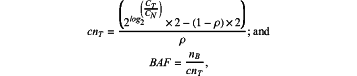 wherein for each of the baseline sample and the tumor sample, cnT represents a CN of a single probe for a test sample CT represents a coverage of the test sample on the single probe, CN represents an average coverage of the baseline sample on the single probe, BAF represents the allele frequency, and nB represents a CN of a non-reference B allele in a germline heterozygous SNP,
wherein for the calculation formulas for the baseline sample, the test sample refers to the baseline sample, and for the calculation formulas for the tumor sample, the test sample refers to the tumor sample;
according to cnT and BAF values of all the probes for the SNP-panel for each chromosome arm included in the regions other than the centromeres and the telomeres for each of the tumor sample and the baseline sample, using a circular segmentation (CBS) method to perform a first segmentation based on cnT so that each chromosome arm is segmented into regions with equivalent CNs; using the CBS method to further segment the regions with the equivalent CNs based on BAF into regions with equivalent allele-specific CNs, and acquiring CNs nB and nA of each genotype of each region of the regions with the equivalent allele-specific CNs, wherein nA is a CN of reference A allele in the germline heterozygous SNP; and for each region, counting cases where nB is equal to 0 and nA is not equal to 0 to obtain the AI scores of the regions other than the centromeres and the telomeres (auto-AI-scores); and
b) the AI scores of the telomeric regions are calculated as follows:
using the pre-processed WGS data of the tumor sample telomeric regions and subtelomeric regions, identifying CNVs within the telomeric regions and subtelomeric regions and dividing the human reference genome into regions with CNs of 0, 1, 2, 3, 4, 5, or 6, within the telomeric regions and subtelomeric regions and evaluating a ploidy P of the tumor sample; determining regions meeting all the following criteria:
I) CN of 1, 3, or 5;
II) CN not equal to the ploidy P; and
III) a region location not spanning across the centromeres; and
counting the regions meeting the three criteria I, II, and III within each telomeric region and subtelomeric region to obtain the AI scores of the telomeric regions (TELO-AI-scores);
5) Using the pre-processed WGS data of the tumor sample and the baseline sample to obtain remaining sequences of regions in the tumor sample other than the telomeric regions and subtelomeric regions obtained in step 4b; and aligning the remaining sequences with the human reference genome, adding with the CNs determined for the telomeric regions and subtelomeric regions of the tumor sample in step 4b to obtain a CN of the tumor sample at the whole genome scale;
6) Using the results in step 5 to determine large-scale state transition (LST) scores,
wherein the LST scores are calculated as follows: for each chromosome arm, according to the CNs of the telomeric regions on each chromosome arm calculated to identify CNVs, counting regions meeting all the following conditions: I) having continuous CNV regions, wherein the continuous CNV regions are adjacent genomic region intervals having the same CNs, and the telomeric regions have a continuous CNV across the entire region; II) having a distance among regions within a first specified threshold; and III) having a length of a region within a second specified threshold; and recording a resulting value as an LST-score for each chromosome arm; and
7) Obtaining a score for the genomic structural variation (STV-score) by calculating a weighted or unweighted sum of at least one selected from the group consisting of the TELO-AI-scores obtained in step 4b, the auto-AI-scores obtained in step 4a, and the LST-scores obtained in step 6;
8) Determining the mutational signature of HRR genes included in the processed NGS sequences from the targeted sequence data for the tumor sample based on statistics of targeted sequencing data of HRR genes obtained from the alignment results of the processed NGS sequences of the tumor sample and the baseline sample in step 2, comprising the following steps:
a) identifying single-nucleotide variants (SNVs) and annotating at a genetic level to obtain annotation results;
b) screening according to the annotation results; and
c) analyzing the mutational signature; and
9) Determining the pathogenic point mutation or the Indel mutation of the HRR genes through the targeted sequencing data of the HRR genes obtained from the alignment results of the processed NGS sequences of the tumor sample and the baseline sample in step 2; determining the biallelic pathogenic mutation burden of the HRR genes based on a number of mutated alleles of each HHR gene; and determining the CN burden of the HRR genes by calculating a number of HRR genes in which the CNVs of the HRR genes exceed a threshold.
|